
Regulatory Frameworks for Bioartificial Organs
A Comparative Analysis of Global Jurisdictions

Abstract
The development of bioartificial organs—hybrid systems integrating living biological materials with bioengineered matrices (e.g., 3D-printed polymers, synthetic hydrogels, or decellularized animal tissues) or technological components (e.g., vascular pumps)—presents a complex regulatory landscape. This overview examines the legal frameworks governing these technologies in the European Union, the United States, Dubai (UAE), China, and Japan, focusing on classification, clinical authorization, and ethical consent requirements.
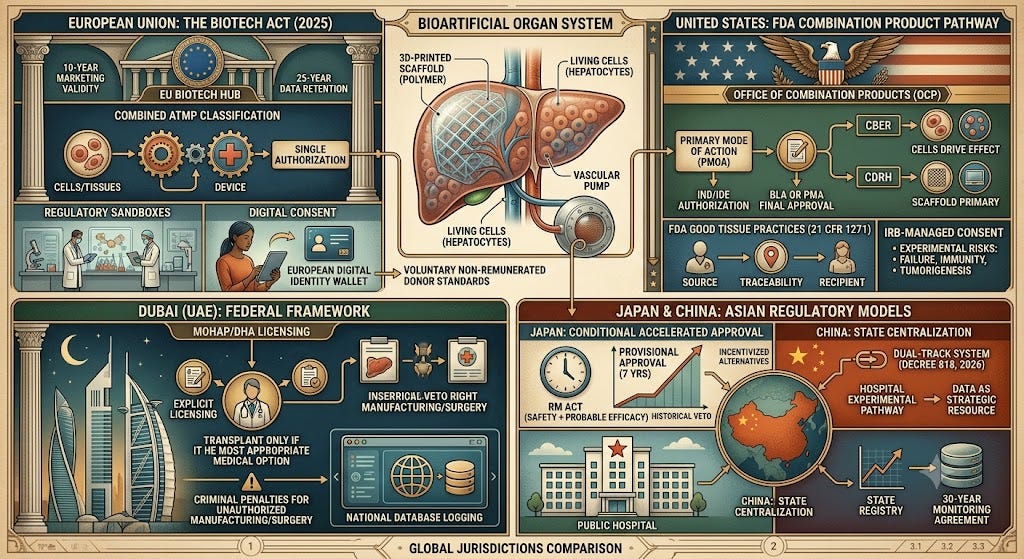
1. European Union: The Biotech Act and Regulatory Convergence
The European Biotech Act, presented in late 2025 and currently under evaluation by the European Parliament and Council, aims to modernize and streamline the EU's regulatory framework to reduce fragmentation and accelerate the market launch of innovative therapies. While the Act does not introduce a new, isolated textual definition for bioartificial organs, it relies on legislative convergence and simplification.
1.1 Classification and Regulatory Mechanisms
Tissue-Engineered Products: Under the amended Regulation (EC) No 1394/2007, if the bioartificial organ contains or consists of engineered cells or tissues, it is classified as a tissue-engineered product or a somatic cell therapy product.
Combined Advanced Therapy Medicinal Products (ATMPs): Because bioartificial organs combine living biological components with a technological framework, the Act coordinates the definition of ATMPs with the Medical Device Regulation (MDR 2017/745). The entire system is regulated as a single ATMP to avoid the need for separate authorizations.
EU Biotech Hub: The organ is evaluated centrally through the new European Biotech Hub, which manages the tissue component and the structural scaffold within a single coordinated framework.
1.2 Regulatory Processes and Safeguards
Regulatory Sandboxes: Manufacturers can test 3D bio-printing and cell culture processes within a controlled environment under the supervision of competent authorities to identify safety requirements.
Clinical Trial Fast-Track: Human trials are accessed via fast-track channels, reducing approval wait times and allowing exemptions from environmental impact testing if the modified cells pose negligible risk.
Marketing Authorization: Marketing authorization for products containing Genetically Modified Microorganisms (GMMs) or modified cells is valid for 10 years and serves as a safeguard clause.
1.3 Patient and Donor Consent
Digital Consent: The Act permits remote informed consent via the EU’s Electronic Identification, Authentication and Trust Services (eIDAS) tools or the European Digital Identity Wallet, ensuring such use remains strictly voluntary.
Data Privacy: Patient consent is divided into separate buckets: the medical procedure and data processing. Data collected under ‘public interest’ or ‘legal obligation’ cannot be deleted upon withdrawal of consent and must be archived for a 25-year minimum retention period.
Accountability: The consent form must include a map of accountability, stating liability for synthetic scaffold failure versus surgical error.
Donor Standards: Allogeneic donor consent must be voluntary, explicit, and non-remunerated, upholding the principle against commercializing the human body.
2. United States: The FDA Combination Product Pathway
Unlike traditional transplants, bioartificial organs are strictly regulated by the FDA as commercial medical products.
2.1 Oversight and Classification
Combination Products: These are overseen by the Office of Combination Products (OCP).
Primary Mode of Action (PMOA): Depending on the PMOA, oversight is assigned to either the CBER (if living cells drive the effect) or the CDRH (if the structural scaffold is the primary driver).
Cell Standards: Products must adhere to 21 CFR Part 1271 (Good Tissue Practices), which requires full traceability from the cell source to the recipient.
2.2 Approval Pathways and Consent
Authorization: Products must undergo preclinical testing, followed by IND or IDE authorization, and ultimately receive final approval via a BLA or PMA.
Clinical Consent: Managed by an Institutional Review Board (IRB), consent requires explicit disclosure of experimental risks, including device failure, immune responses, and tumorigenesis.
Surgical Exemptions: The ‘Same Surgical Procedure’ exemption allows for minimal processing of autologous cells during the same operation without full manufacturing oversight; however, external culturing or 3D printing triggers full Section 351 Biologic oversight.
3. Asian Regulatory Models: Dubai, China, and Japan
3.1 Dubai (UAE): The Centralized Federal Framework
Federal Decree-Law No. 15 of 2025: The UAE explicitly recognizes and regulates manufactured organs. Operations require explicit licensing from MOHAP or DHA. A transplant is permitted only if it represents the most appropriate medical option, and all manufactured organs must be logged in a centralized national database. Unauthorized manufacturing or unlicensed surgeries carry severe criminal penalties.
3.2 Japan: Conditional Accelerated Approval
Japan prioritizes speed via the Act on the Safety of Regenerative Medicine (RM Act). Products can obtain provisional approval (up to 7 years) and be marketed as soon as safety and ‘probable’ efficacy are demonstrated. Japan uses an opt-in model in which the next of kin retains the legal right to veto organ harvesting, which has historically incentivized the development of artificial alternatives.
3.3 China: State Centralization
Regulated by the NMPA, China adopts a ‘Dual-Track’ system under Decree No. 818 of 2026. Top-tier public hospitals can bypass lengthy clinical procedures for experimental transplants. Clinical and biological data are deemed ‘strategic national resources’ and cannot leave the country. Recipients must consent to long-term monitoring, agreeing that data will be kept in a State registry for at least 30 years.
References
Europe
European Union European Biotech Act: The proposal (COM(2025) 1022 final) aims to modernize EU biomanufacturing and research processes.
European Biotech Act: EU Legislation in Progress
Precision for Medicine: EU Biotech Act Overview
United States
21 CFR Part 1271: This regulation governs the manufacturing and handling of Human Cells, Tissues, and Cellular and Tissue-Based Products (HCT/Ps).
Electronic Code of Federal Regulations (eCFR): 21 CFR Part 1271
United Arab Emirates
Federal Decree-Law No. (15) of 2025: This law, effective January 1, 2026, expands the national framework to include non-human and manufactured organs.
UAE Ministry of Justice Legislation Portal
Tamimi & Co: Legislative Analysis of Decree-Law No. 15 of 2025
China
Decree No. 818 of 2026, formally titled the Regulations on the Administration of Clinical Research and Clinical Translation and Application of Biomedical New Technologies, establishes a dual-track pathway for biomedical innovation.
Cisema: China Issues Landmark Regulation on Biomedical New Technologies
Morgan Lewis: China’s Order 818 Analysis
Japan
Act on the Safety of Regenerative Medicine (Act No. 85 of 2013): This Act provides the legal basis for the rapid provision and safety of regenerative medicine.
Japanese Law Translation: Act on the Safety of Regenerative Medicine
 | A guest post by
|